

WHAT IS MYASTHENIA GRAVIS?
While the annual risk of myasthenia gravis disease is 8-10 people per 1 million, the number of diagnosed patients is estimated to be 150-250 people per 1 million. The disease can occur in any race and at any age. However, it is more common in women than in men.
Antibodies detected in myasthenia gravis disease
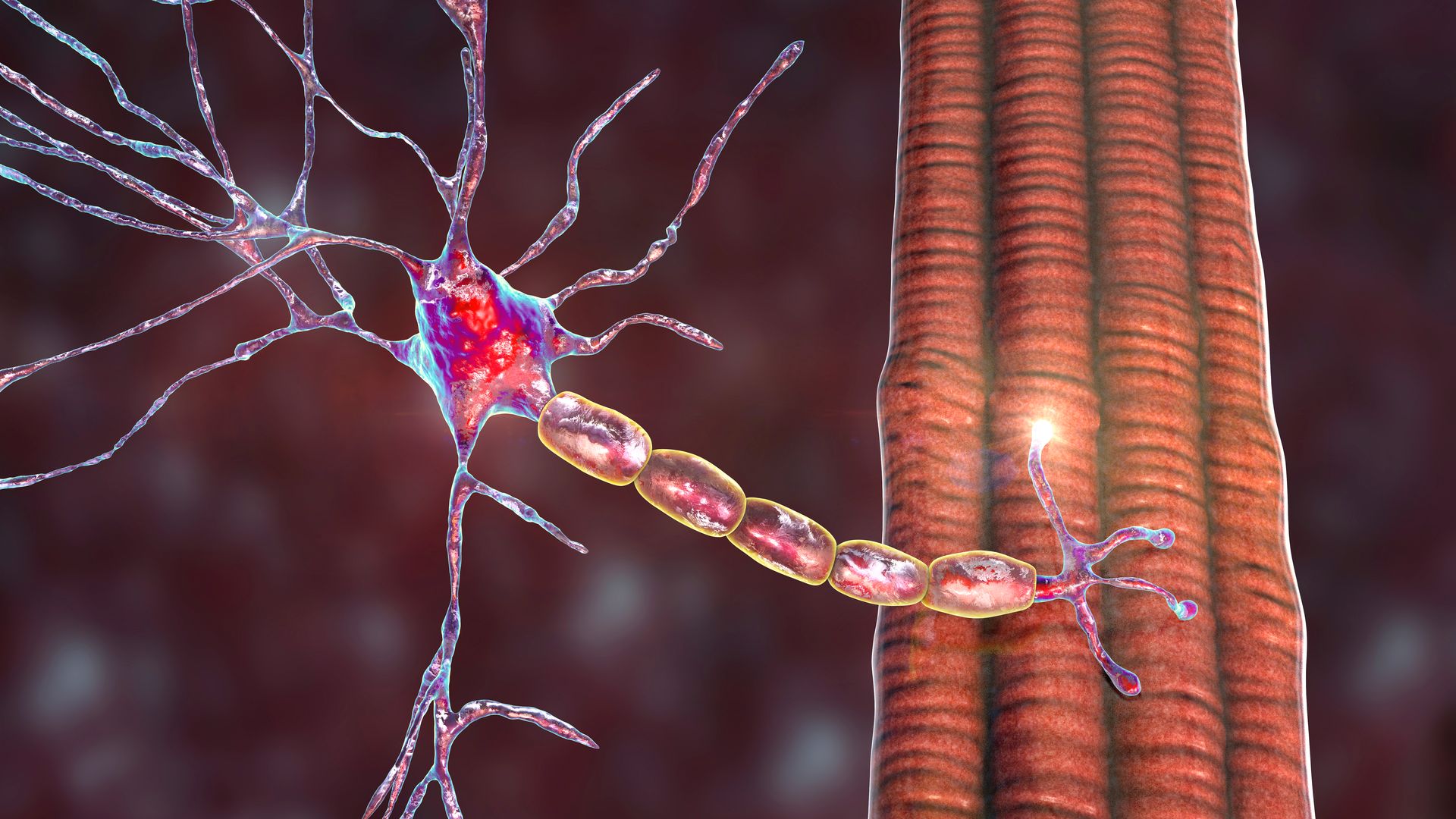
In order to make a movement, the brain sends an electrical signal to the muscle via motor nerves, this electrical signal turns into a chemical signal at the muscle nerve junction and causes acetylcholine (ACh) molecules to combine with acetylcholine receptors and the formation of a muscle action potential, and the muscle fiber contracts.
Various studies have shown that AChR antibodies are detected in 67-93% of myasthenia gravis patients. In various studies, approximately 30-40% of AChR antibody negative patients are found to be MuSK (Muscle specific kinase) positive.
Antibodies against some other components of the muscle-nerve connection, such as LRP4 (Lipoprotein receptor 4), are also thought to play a role in the development of MG. While AChR antibodies are fairly well characterized, information about other antibodies is evolving over time.
Symptoms of myasthenia gravis disease
The most important feature of myasthenia gravis is a fluctuating muscle weakness that increases with fatigue and partially improves with rest. It is quite typical for patients to wake up well-rested in the morning and complain of muscle weakness that gradually increases throughout the day. A muscle weakness involving the striated muscles is observed. The disease most often begins with unilateral or bilateral, and when bilateral, asymmetric eyelid ptosis and double vision. Difficulty in swallowing and chewing, difficulty in speaking, and weakness in arm and leg muscles may soon be added to these symptoms. Symptoms may manifest themselves as difficulty climbing hills or stairs, difficulty lifting the arms up, or the inability to lift one or two fingers for a while.
The most severe form of the disease is respiratory difficulty that occurs when the respiratory muscles are affected. In these patients, weakness in the tongue, soft palate and chewing muscles, and nasal speech are noted. In seriously ill patients, it becomes difficult to swallow food, drinking water and liquids come through the nose, and nutrition is only possible through the nasogastric tube. Infections, excessive fatigue and heavy stress often negatively affect the course of the disease.
Diagnosis of myasthenia gravis disease
Diagnosis is made by the presence of typical clinical symptoms and the presence of AChR antibody or MuSK antibody. In addition to antibody tests, electromyography (EMG) is important in the diagnosis of myasthenia gravis. Here, two important methods come to the fore: The first is the sequential nerve stimulation test. As a result of this test, detecting a gradient of 10% or more with 3 Hz stimulation is a finding that supports the diagnosis. The second method is single fiber EMG examination. Increased jitter response and detection of blocks are features seen in MG.
Edrophonium (Tensilon) and Neostigmin methylsulfate (Prostigmin) are other tests used in diagnosis. The effects of these drugs appear in a few minutes and last for a few minutes. The response to the Edrophonium or Neostigmine test is considered positive when there is a significant improvement in the patient's current symptoms and examination findings.
Thymus gland in myasthenia gravis patients
A tumor of the thymus gland called thymoma is detected in approximately 10% of MG patients. The thymus gland is a tissue that shrinks with age. However, in myasthenic patients, on the contrary, enlargement of the thymus gland and thymoma may be detected in a small group of patients. For this reason, non-contrast thorax computed tomography examination should be performed in every patient who is considered to be diagnosed with MG. Thymectomy is recommended especially in young and AChR antibody positive MG patients.
Myasthenia gravis treatment
The first step in treatment is the administration of pyridostigmine, which prevents the destruction of acetylcholine at the muscle-nerve junction. This drug is symptomatic and its duration of action varies from patient to patient. However, its effect lasts for an average of 4-6 hours. Pyridostigmine should be part of the initial treatment for most patients with myasthenia gravis, with the dose adjusted according to symptoms. Corticosteroids or immunosuppressive drug treatments are also used in myasthenia gravis patients. The corticosteroid dose can be increased up to 1mg/kg, but it should be kept in mind that steroids may have a risk of worsening symptoms within 7-10 days. In order to protect the patient from this effect, it would be appropriate to start the steroid dose low and gradually increase it, or if it is necessary to start at a high dose, hospitalization and close follow-up of the patient is appropriate. When corticosteroids cannot be used, a non-steroidal immunosuppressive agent should be used. These agents include azathioprine, cyclosporine, mycophenolate mofetil, methotrexate, and tacrolimus. In patients with treatment-resistant myasthenia gravis, treatments such as chronic intravenous immunoglobulin (IVIg) and chronic plasmapheresis (plasma exchange - PLEX), cyclophosphamide, rituximab, and eculizumab can be used.
Myasthenic crisis
Myasthenic worsening that occurs with respiratory distress and requires respiratory support (mechanical ventilation) is called myasthenic crisis. It requires intensive care admission and aggressive management of the treatment. Plasmapheresis and IVIg are used as short-term treatment for impending and emerging myasthenic crisis in patients with significant breathing or swallowing difficulties. Corticosteroids or other immunosuppressive agents are often initiated simultaneously to achieve a sustained clinical response.
Although clinical studies suggest that IVIg and plasmapheresis are equally effective in treating impending or emerging myasthenic crisis, expert consensus suggests that plasmapheresis is more effective and works faster. The choice between the two treatments depends specifically on the patient's comorbidities and several other factors, including the accessibility of these treatments.
Resources
- Sanders D.B.; Wolfe, G.I.; Narayanaswami, P., “Developing treatment guidelines for myasthenia gravis,” Annals of the New York Academy of Sciences, Ocak 2018, 1412(1): 95-101.
- Sanders D.B.; Wolfe, G.I.; Benatar, M.; Evoli, A.; Gilhus, N.E.; Illa, I.; Kuntz, N.; Massey, J.M.; Melms, A.; Murai, H.; Nicolle, M.; Palace, J.; Richman, D.P.; Verschuuren, J.J.G.M.; Narayanaswami, P., “International consensus guidance for management of myasthenia gravis: Executive summary,” Neurology, 26 Temmuz 2016, 87(4): 419-425.
- Güngör-Tunçer, Ö., “MuSK antikor pozitif, MuSK antikor negatif ve seropozitif myastenia gravis’te klinik, laboratuvar ve tedavi yanıtlarının karşılaştırması,” İstanbul Üniversitesi Tıp Fakültesi Nöroloji Bölümü uzmanlık tezi, 2005.
- Güngör-Tunçer, Ö., “Anti-MuSK antikor pozitif Myasthenia Gravis hastalarında ardışık sinir uyarım testi ile yüz ve ekstremite kaslarının yanıtlarının değerlendirilmesi,” İstanbul Üniversitesi, DETAE Sinir Bilim Elektro-Nöro-Fizyoloji Bölümü, yüksek lisans tezi, 2013.
- Deymeer, F.; Güngör-Tunçer, Ö.; Yılmaz, V.; Parman, Y.; Serdaroğlu, P.; Özdemir, C.; Vincent, A.; Saruhan-Direskeneli, G., “Clinical comparison of anti-MuSK vs anti-AChR-positive and seronegative myasthenia gravis,” Neurology, 20 Şubat 2007, 68(8): 609-611.
- Nagappa, M.; Mahadevan, A.; Gangadhar, Y.; Patil, S.A.; Bokolia, S.; Bindu, P.S.; Sinha, S.; Taly, A.B., “Autoantibodies in acquired myasthenia gravis: Clinical phenotype and immunological correlation,” Acta Neurologica Scandinavica, Mayıs 2019, 139(5): 428-437.
- Jaretzki, A.; Barohn, R.J.; Ernstoff, R.M.; Kaminski, H.J.; Keesey, J.C.; Penn, A.S.; Sanders, D.B., “Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America,” Neurology, 12 Temmuz 2000, 55(1): 16-23.
- Ozdemir C, Young RR. The results to be expected from electrical testing in the diagnosis of myasthenia gravis. Ann N Y Acad Sci. 1976;274:203-22. doi: 10.1111/j.1749-6632.1976.tb47686.x. PMID: 1066987.
© 2026 made by AHMED AKIN
The information contained on the website is to provide support. It is not a substitute for the physician examining the patient for medical purposes and making a diagnosis.
CommentPlease write to us to make comment